Research Article
Chromium Isotopes Detection in their Ores with Minimal Errors

Loai Aljerf1* and Nuha AlMasri2
1Department of Basic Sciences, Faculty of Dental Medicine, Damascus University, Mazzeh Highway, Almazzeh, Damascus, Syria
2Department of Chemistry, Faculty of Medicine, Syrian Private University, Damascus, Syria
*Address for Correspondence: Loai Aljerf, Department of Basic Sciences, Faculty of Dental Medicine, Damascus University, Mazzeh Highway, Almazzeh, Damascus, Syria, Tel: +963-93 34 46 993; Email: [email protected]; [email protected]
Dates: Submitted: 21 July 2018; Approved: 03 September 2018; Published: 04 September 2018
How to cite this article: Aljerf L, AlMasri N. Chromium Isotopes Detection in their Ores with Minimal Errors. Ann Adv Chem. 2018; 2: 045-054. DOI: 10.29328/journal.aac.1001013
Copyright License: © 2018 Aljerf L, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Chromium; Abundance; Isotope; Mass spectroscopy; Precision

Abstract
The industrial production and use of chromium have grown considerably during the past five decades. Abundances of the chromium isotopes in terrestrial samples are identical to 0.01%. Among the dominant species of chromium, the trivalent form widely occurs in nature in chromite ores which is extremely immobilized especially in water bodies. Samples were mixtures of separated chromium isotopes and the calibration was made with the same species as those used in the measurements. The method had simplified the conversion of the ores to chromyl fluoride since the element could be readily separated as lead chromate from the leaching of chromite-sodium peroxide fusions. Isotope assay of chromyl fluoride under certain conditions was measured and the measurements of chromium isotopic anomalies ratios and isotope abundance of the chromite ores have been assessed. These provided sufficient quantitative mass spectrometric data, which were analyzed to calculate the abundance and the mean atomic mass of the questioned isotopes. Based on the high mass spectroscopy stability and the correction factors, the results were of good precision (incl. negligible systematic errors normally associated to inter-laboratory discrepancies) and the Cr isotopes availability (52Cr > 53Cr > 50Cr > 54Cr) was in conjunction with other classical tools such as oxygen isotopes. This paper is important for paleoecological, environmental, archeological, forensic, and nuclear researchers.
Introduction
Chromium is one of the elements on which no systematic study of the isotopic abundances has been made with regard to the geological source of the materials [1,2]. All the results of previously reported works were obtained with chemical reagents taken from the usual stores of these materials. As is evident in table 1, there is no real agreement among any of the results.
| Table 1: Previous determinations of the abundances of the chromium isotopes, in atoms percent. | ||||
| Measurement
Ion source reagent observer |
1 Gas discharge Cr(CO)6 [3] |
2 Thermionic Cr metal [4] |
3 Electron impact CrCl3 [5] |
4 Electron impact CrCl3 [6] |
| Isotope 50 | 4.9 | 4.49 | 4.31±0.04 | 4.41±0.06 |
| Isotope 52 | 81.6 | 83.8 | 83.8±0.14 | 83.5±0.11 |
| Isotope 53 | 10.4 | 9.55 | 9.55±0.09 | 9.54±0.06 |
| Isotope 54 | 3.1 | 2.31 | 2.38±0.02 | 2.61±0.09 |
The main supply of chromium chemicals available in many countries, is from the chromite deposits [7,8]. Thus, it appears that the variation of the results listed in table 1, with the exception of those of Nowak [3], may be due to fractionation brought about in the chemical processing of the ores or by natural geological processes; or, they may be due to unknown discriminations involved in the various instruments with which the measurements were made.
With this in mind, a study was made of the abundances of the Cr isotopes in samples of chromite and other chromium bearing minerals collected from well-characterized deposits. The present paper reports the results of the initial studies dealing with the primary mineral, chromite. Descriptions are given of the precautions taken to eliminate instrumental discriminations and the development of a technique by which highly precise measurements can be made with this and the other transition elements. A report of the studies on secondary chromium minerals will be given in a subsequent publication.
Materials and Methods
Materials
Survey of the chromite sources production in the world
Table 2 lists eighteen chromite specimens obtained from countries responsible for 81% of the chromite ore produced from 1931 to 2018 [9-22].
| Table 2: Source and type of chromite used in the study. | |||
| Ore number | Location of deposit | Remarks | References |
| 1 | Tiebaghi, New Caledonia | Red ore | [9] |
| 2 | Fantoche, New Caledonia | Grey ore | [9] |
| 3 | Acoje or Zambales, Philippines | [10] | |
| 4 | Rustenberg, Western Transvaal | [11] | |
| 5 | Lydenberg, Eastern Transvaal | [11] | |
| 6 | Western Transvaal | Exact location unspecified | [11] |
| 7 | Transvaal | [11] | |
| 8 | Rhodesia | Great Dyke, 190 miles NNE of Selukwe, concentrates from alluvial deposits | [12] |
| 9 | Rhodesia | Great Dyke, 150 miles NNE of Selukwe, friable lump | [12] |
| 10 | Elazig Province, Turkey | Guleman ore from near Erganimaden | [13] |
| 11 | Ural Mountains, Russia | Russian chrome, several possible locations | [14,15] |
| 12 | Angelita, Cuba | Concentrates of Holquin ore | [16,17] |
| 13 | Black Lake, Quebec | Recent operation | [18] |
| 14 | Black Lake, Quebec | Concentrates | [18] |
| 15 | Black Lake, Quebec | Old sample from early operations at Black Lake | [18] |
| 16 | Hartford County, Maryland | Near Cooktown | [19] |
| 17 | Texas, Pennsylvania | [20] | |
| 18 | Benbow, Montana | Concentrate | [21,22] |
Methods
Strategy: In a previous publication [23] a preparation of chromyl fluoride (CrO2F2) by vacuum distillation from a solid-solid mixture of chromium trioxide (CrO3), and cobalt (iii) fluoride (CoF3), was described. The high vapor pressure of chromyl fluoride and its convenient preparation made this compound ideal as a means of introducing the element into the mass spectrometer (MS) as a gas, which affords the experimenter the advantage of extremely stable sample conditions. To avoid any possibility of introducing errors due to the possible isotopic effects on the fragmentation of this compound, CrO2F2+ ions were chosen as the ion currents on which to base each abundance determination [24]. The anisotopic nature of fluorine simplified the handling of the MS data.
Analytical protocols: Since considerable chemical manipulation is required to convert chromite ores (Fe, Mg)Cr2O4 to CrO3, a simpler procedure for preparing CrO2F2 was sought. In the course of this search, experiments revealed that the CrO3 used in the above method could be replaced by lead chromate (PbCrO4). This greatly simplified the conversion of the ores to chromyl fluoride, since the element could be readily separated as lead chromate from the leaching of chromite (Cr2O42-)-sodium peroxide (Na2O2) fusions. To accomplish this, the ores were finely pulverized, fused with an excess of sodium peroxide, and leached with distilled water. The leaching was neutralized with glacial acetic acid, filtered to remove the silica, and then solid lead acetate was added to precipitate the chromium as lead chromate. Excess reagents were used throughout to ensure complete separation of the chromium, thus avoiding any possible isotopic fractionation effects during chemical treatment. The lead chromate was filtered, washed, air dried, and then ignited at from 400 to 500°C [25,26] to remove any residual organic matter as well as to dry the material. The ignited lead chromate was immediately transferred into a dry box through the route revealed in figure 1, where it was stored until used.
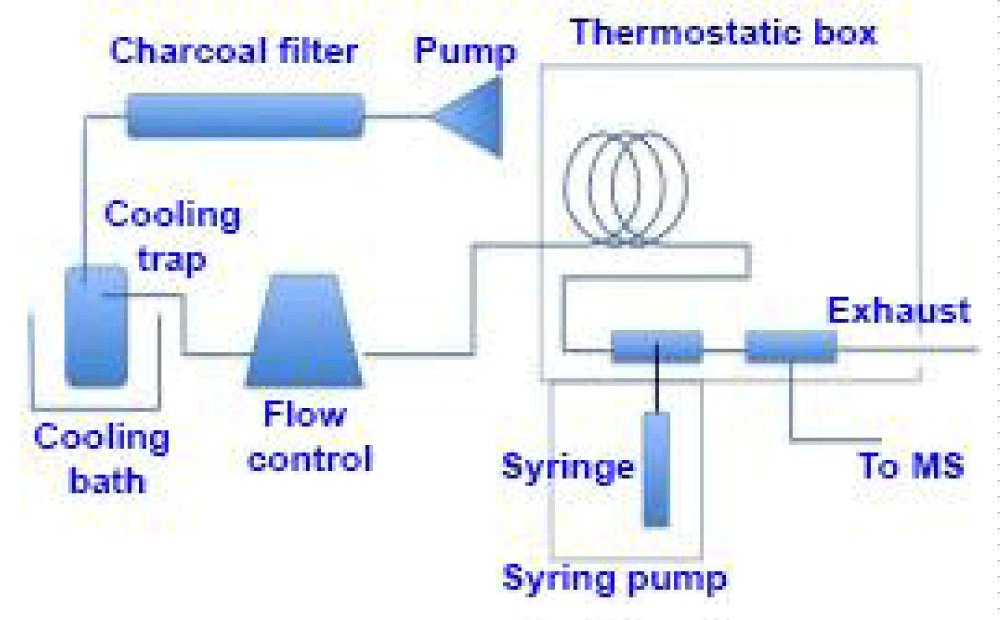
Figure 1: Schematic view of the novel direct aqueous injection system.
For conversion to chromyl fluoride the lead chromate was finely ground and intimately mixed with an excess of CoF3. The mixture was placed in a copper reaction tube. Bronze turnings were placed above the charge to prevent blow-over of the reactants during subsequent evacuation of the reaction tube.
The reaction tube was removed from the dry box and attached to the remainder of the sample preparation apparatus. The assembled sample preparation apparatus consisted of a 0.25 in. copper reaction tube which was about 6 in. long, a 0.25 in. copper U-tube, and a packless, bellows-type valve (Hoke M482). The components were joined by means of standard flare fittings, and the assembled apparatus was attached to the inlet system of the mass spectrometer through a stainless steel fitting (Hoke S24). An aluminum gasket was used to make the latter connection vacuum tight.
MS prepreparation method: A furnace was placed about the reaction tube and the temperature was raised to 375°C. This initial heat served to outgas the reactants and reaction tube without any loss of chromyl fluoride since the fluorination reaction does not proceed at this temperature. The outgassing was accompanied by an increase of pressure which was monitored by the inlet vacuum system Pirani gauge. When the pressure had returned to near normal, a dry-ice-trichloroethylene (C2HCl3) slush was placed about the U-tube to trap the chromyl fluoride, and the furnace temperature was increased to 550°C. Again the pressure in the inlet vacuum system increased due to oxygen produced during the fluorination of the lead chromate. When the pressure had returned to normal, the reaction was considered complete. The furnace was removed, and the chromyl fluoride was then treated like any other condensable gas. Samples of this compound could be stored in the copper container for several days without any apparent deterioration.
MS optimization method: The mass spectrometer employed in the study was a 180° instrument of 5 in. radius (Consolidated Electrodynamics Corporation, Model 21-220, modified in our laboratory). A 200 µA current of 70 V electrons ionized the chromyl fluoride at an analyzer pressure of less than 5 x 10-7 mm Hg, indicated by an ionization gauge placed within 12 in. of the ion source. The vacuum system was arranged to give differential pumping. Preliminary experiments to determine the optimum instrumental operating conditions were performed using a reference sample of chromyl fluoride prepared from lead chromate, which had been prepared from reagent-grade chromic bromide (Br3Cr). This chromyl fluoride was assayed over the complete mass range of the instrument and found to be 99+ percent pure with the principal impurity being HF.
Results and Discussion
MS optimization outcomes
Preliminary experiments showed that the sensitivity of the MS for the CrO2F2+ ions was constant in the ion accelerating voltage range of from 1600 to 1700 V. This meant that the isotopic CrO2F2+ ion currents could be voltage scanned in that range, which was more convenient than magnetic scanning. It was found that the most reproducible results were achieved when the CrO2F2+ ions were manually scanned to obtain the desired mass spectrograms. Ten spectrograms were made by scanning the mass region 120-124 in alternate directions. Consecutive spectrograms were averaged, which resulted in nine sets of data for each assay of any particular chromyl fluoride sample.
Oxygen isotopes intervention and their correction
The oxygen isotopes could not be ignored in any isotope determination using CrO2F2+ ions, since ion currents at masses 121, 125 and 126 were observed and their magnitudes agreed with those calculated for ions containing 17O and 18O.
Since direct measurement of the oxygen isotopes in CrO2F2 was not possible, an indirect measurement was made. Given that the production of chromyl fluoride involved the fluorination of PbCrO4 by means of CoF3, some means of examining directly the abundance of the oxygen isotopes in this chromate was sought. The most direct procedure developed involved the reaction of lead chromate with potassium fluobromite, KBrF4 [26,27].
The liberated molecular oxygen was introduced directly into a dual collector, 60°, 6 in. radius mass spectrometer and the 34O+/32O+ and 33O2/32O2 ratios measured. Atmospheric oxygen, for which Nier [4] and Young et al. [28] gave an absolute ratio for 34O2/32O2 of 0.00409, was used as a comparison standard in order to place all measurements on an absolute basis.
All of the PbCrO4 samples prepared from the ores listed in table 2 and the distilled water were examined. The results of the KBrF4 fluorinations along with that of CoF3 are summarized in table 3.
| Table 3: KBrF4 and CoF3 fluorination reactions. | |||
| Item | Reaction | 33O2/32O2 | 34O2/32O2 |
| 1 | PbCrO4+KBrF4 O2+. ..... | 0.000730 ± 5 | 0.003930 ± 5 |
| 2 | H2O+KBrF4 O2+...... | 0.000730 ± 5 | 0.003930 ± 5 |
| 3 | PbCrO4+CoF3 O2+...... | 0.000730 ± 5 | 0.003930 ± 5 |
From these results, it appeared that the oxygen in the PbCrO4 was derived primarily from the distilled water used to leach the sodium peroxide-chromite fusions and to wash the precipitated lead chromate. To ensure that, fractionation of the oxygen isotopes did not occur during the fluorinations. One experiment was performed in which four different samples of O2 were collected at various times during the reaction. Assay of this gas gave identical results with those already cited.
An exchange experiment designed to confirm the nature of the exchange between CrO42- and H2O8 was performed. Vacuum-dried sodium chromate was dissolved in water enriched in 18O to 5 atom percent. As soon as complete solution was achieved, the water was distilled off and crystalline Na2CrO4 recovered.
After being dried under vacuum at 70°C for 7 hr, this material was fluorinated with KBrF4. (In this case the fluorination proceeded smoothly at 200°C.) Measurement of the 34O2/32O2 ratio for the evolved oxygen indicated complete exchange within the short time of the experiment. This is essentially in agreement with the results of Brodskii and Dontsova [29] and of Mills [30] whose measurements of the oxygen isotopes were based on the density of the water employed. On the basis of these results, it was concluded that correction of the observed CrO2F2+ ion currents for the oxygen isotopes had to be based upon the observed abundances in the distilled water. Accordingly, corrections of the observed ion currents were made on the basis of 16O/17O = 2740, and 16O/18O = 508.9 for the oxygen isotopes in the distilled water and the assumption that the oxygen in chromyl fluoride was a random collection of oxygen atoms.
Thus, the calculated abundances of the 16O, 16O; 16O, 17O; and 16O, 18O combinations were 99.536, 0.073, and 0.391 percent, respectively. Any other combinations were present to less than 0.001 percent and were considered negligible.
The equations which relate the observed ion currents to the abundances of the chromium isotopes are:
120M = 50A (1)
122M – 0.00393 50A = 52A (2)
123M – 0.00073 52A = 53A (3)
124M – 0.00073 53A – 0.00393 52A = 54A (4)
Where M is the observed ion current, A is the relative abundance, and the superscript is the isotopic mass number. The factors 0.00073 and 0.00393 are the ratios 16O, 17O/16O, 16O and 16O, 18O/16O, 16O, respectively. A simplification which introduced negligible error was the replacement of the terms 52A and 53A in the left hand members of the equations (3) and (4) with 122M and 123M, because the products of the decimal fractions involved were negligible. Thus equations (1) to (4) were reduced to:
122M – 0.00393 120A = 52A (5)
123M – 0.00073 122A = 53A (6)
124M – 0.00073 123A – 0.00393 122A = 54A (7)
These were used in all the subsequent treatment of the data.
Experimental pressure effect
Since there was small probability of obtaining equal ion currents for all eighteen samples of chromyl fluoride, the reference sample of chromyl fluoride was assayed at one-half, at normal, and at twice the normal operating pressure to determine whether or not the assays were pressure dependent. The results of this experiment are shown in table 4. They indicate that no pressure effect on the assays occurred and that the electrometer tube input resistor was truly ohmic in its behavior.
| Table 4: Isotope assay of chromyl fluoride at various pressures (Isotope abundance, atoms percent). | ||||
| Isotope | 50 | 52 | 53 | 54 |
| Half normal pressure | 4.335 | 83.768 | 9.529 | 2.368 |
| Normal pressure | 4.332 | 83.759 | 9.535 | 2.373 |
| Twice normal pressure | 4.343 | 83.760 | 9.524 | 2.372 |
Comparative inter-laboratory study
Comparison of the assays of the chromyl fluoride prepared from the eighteen chromite ores depended on the stability of the instrument during the period required to assay the materials and the absence of any fractionation effects during the production of the CrO2F2 and its subsequent volatilization into the inlet system of the mass spectrometer. Accordingly, three different check experiments were devised. The first check involved chromyl fluoride made from commercial CrO3 and assayed four different times during a 12 day period. The second and third checks involved different chromyl fluoride preparations from lead chromate which had been made from reagent-grade chromic bromide. In the second check, two samples of chromyl fluoride, made from different portions of the lead chromate, were assayed 5 days apart. In the third check, an additional preparation was assayed four times during a 5 day period. Thus, all fractionation or discrimination possibilities were considered. The average assay and single standard deviation of each check are shown in table 5 in columns A, B and C, respectively. These results indicated that comparison of the assays of the eighteen ore samples could be made with no concern about day-to-day instrumental drift or chemical fractionation effects.
| Table 5: Instrument stability (Isotope abundance, atoms percent). | |||
| Mass | A | B | C |
| 50 | 4.347 ± 0.005 | 4.353 ± 0.005 | 4.347 ± 0.003 |
| 52 | 83.754 ± 0.016 | 83.760 ± 0.008 | 83.760 ± 0.005 |
| 53 | 9.515 ± 0.009 | 9.515 ± 0.007 | 9.521 ± 0.005 |
| 54 | 2.384 ± 0.005 | 2.372 ± 0.003 | 2.372 ± 0.004 |
| Variations cited are standard deviations | |||
Analytical challenges
Another potential error involved the exact nature of gas flow in the mass spectrometer. The sample inlet system of the instrument was equipped with a viscous leak which consisted of a 5 in. length of copper capillary tubing of 0.005 in. internal diameter terminated by an adjustable constriction. Differential pumping in the ion source indicated the probability that mass discrimination due to effusive flow was present. However, it has been shown that the nature of the correction factor that should be applied to isotope measurements for this and other instrumental discriminations is dependent upon the detailed physical conditions existing in a particular mass spectrometer [31,32]. It is therefore most reliable to determine these correction factors by calibration of the instrument by means of mixtures of separated isotopes.
Mixtures of separated chromium isotopes were first considered for this calibration. Thus the calibration would have been made with the same species as those used in the measurements. Attempts to use CrO2F2 prepared from mixtures of these materials were fruitless due to an inability to assay the separated isotopic materials satisfactorily with the instrument at hand. This was principally the result of an exchange between the chromium of the chromyl fluoride and the chromium of the Nichrome V (9–21% Cr, 2.5% Mn (max), 1.0% Fe (max), 0.75–1.6% Si, 0.15% C (max), balance Ni) used in the construction of the ion source. This exchange resulted in assay errors whose magnitude depended on the difference of the sample composition from that of normal chromium.
Overcoming solutions
For samples whose isotopic abundances were normal, errors from this effect were computed to be negligible. The difficulty might have been circumvented by constructing a new source of some other material or gold plating the existing source. Both of these possibilities were considered and the latter was seriously contemplated. This will be done for the extension of the work to secondary chromium materials where the preliminary experiments indicate differences in the isotopic constitution.
Suggested corrective measures: A compromise calibration for the present phase of the work was accomplished with a mixture of separated nitrogen isotopes which was carefully prepared and assayed according to the method described by Junk and Svec [33] to produce absolute abundance values. This mixture was then assayed with the mass spectrometer employed throughout the chromium tests under conditions in which the range of ion acceleration voltage was as nearly identical as possible with those under which the chromium assays were made. The value obtained here for the nitrogen isotope mixtures was 42.29 atom percent 15N compared to 42.18 obtained by Junk and Svec [33]. However, the indicated agreement to one part in 423 was obtained only when the data were corrected for fractionation due to effusive gas flow from the ion source. A correction factor equal to the square root of the inverse ratio of the masses involved had to be applied. Because the precision obtained in this calibration approached that obtained with the chromium measurements, it was safe to assume that all of the mass discrimination due to gas flow could be considered to be the result of effusion of the sample material from the ion source.
After these preliminary experiments, the samples of chromyl fluoride prepared from the eighteen chromite ores were assayed along with the reference sample during a 2 week period. The observed data for the ores were first corrected for gas flow discrimination and then for the oxygen isotope effect. The results are listed in tables 6,7. Table 6 gives the individual assays of the eighteen samples and the average assay. Table 7 gives the standard deviation associated with each individual assay and the average of the eighteen individual standard deviations.
| Table 6: Isotope abundance determination of the chromite ores (Isotope abundance, atoms percent). | ||||
| Isotopes Samples | 50 | 52 | 53 | 54 |
| 1 | 4.357 | 83.763 | 9.497 | 2.373 |
| 2 | 4.348 | 83.774 | 9.506 | 2.371 |
| 3 | 4.357 | 83.757 | 9.506 | 2.379 |
| 4 | 4.350 | 83.748 | 9.522 | 2.380 |
| 5 | 4.351 | 83.764 | 9.511 | 2.374 |
| 6 | 4.347 | 83.781 | 9.505 | 2.368 |
| 7 | 4.348 | 83.771 | 9.510 | 2.371 |
| 8 | 4.357 | 83.750 | 9.496 | 2.388 |
| 9 | 4.357 | 83.764 | 9.500 | 2.379 |
| 10 | 4.353 | 83.754 | 9.515 | 2.378 |
| 11 | 4.346 | 83.769 | 9.509 | 2.377 |
| 12 | 4.348 | 83.759 | 9.514 | 2.379 |
| 13 | 4.346 | 83.762 | 9.515 | 2.377 |
| 14 | 4.352 | 83.764 | 9.509 | 2.374 |
| 15 | 4.347 | 83.759 | 9.519 | 2.376 |
| 16 | 4.348 | 83.767 | 9.512 | 2.373 |
| 17 | 4.352 | 83.771 | 9.508 | 2.369 |
| 18 | 4.362 | 83.767 | 9.502 | 2.369 |
| Ave. | 4.352 | 83.764 | 9.509 | 2.375 |
| SD | 0.004 | 0.008 | 0.007 | 0.005 |
| Table 7: Standard deviation associated with each abundance determination of the chromite ores shown in Table 6 (Standard deviation x 1000). | ||||
| Isotopes Samples | 50 | 52 | 53 | 54 |
| 1 | 10.1 | 8.80 | 5.30 | 4.70 |
| 2 | 3.80 | 6.70 | 5.00 | 3.10 |
| 3 | 9.40 | 10.9 | 12.5 | 4.00 |
| 4 | 2.60 | 6.00 | 4.60 | 2.30 |
| 5 | 4.80 | 9.00 | 3.90 | 3.50 |
| 6 | 4.20 | 10.2 | 4.20 | 4.50 |
| 7 | 2.60 | 5.50 | 5.60 | 3.40 |
| 8 | 5.70 | 9.60 | 4.20 | 3.90 |
| 9 | 3.30 | 6.00 | 3.20 | 2.90 |
| 10 | 2.40 | 3.80 | 3.60 | 2.00 |
| 11 | 5.70 | 7.90 | 4.40 | 3.80 |
| 12 | 3.10 | 7.30 | 2.60 | 3.50 |
| 13 | 10.5 | 11.6 | 5.60 | 3.70 |
| 14 | 2.10 | 6.70 | 5.30 | 4.50 |
| 15 | 2.40 | 13.6 | 13.8 | 3.60 |
| 16 | 3.50 | 9.70 | 8.50 | 3.10 |
| 17 | 3.80 | 8.10 | 3.70 | 5.00 |
| 18 | 10.3 | 14.7 | 7.20 | 3.40 |
| Ave. | 5.00 | 8.70 | 5.70 | 3.60 |
Comparison of the results listed in tables 6,7, shows that the standard deviation from the average assay of the eighteen ores was nearly identical to the average standard deviation associated with each individual assay. It must therefore be concluded that there is no variation in the isotopic composition of chromium with respect to the source of chromite ore. In order to examine the statistics of the determinations further, every individual datum from each of the assays was grouped into a composite of 170 items for each isotope abundance.
The standard deviation of the composite data (Table 8) is larger but is more reliable than that of the average assay of the eighteen ores, because in the calculation of the latter, the standard deviation for each individual assay was ignored.
| Table 8: Composite of all data for chromiteores. | |||
| Mass | Abundance (%) | ± Standard deviation | Number of items outside 3 σ limit |
| 50 | 4.352 | 0.008 | 2 |
| 52 | 83.764 | 0.012 | 0 |
| 53 | 9.509 | 0.009 | 0 |
| 54 | 2.375 | 0.006 | 1 |
Despite the good precision of the data above, it was necessary to consider whether or not the values obtained represented the absolute abundances of the chromium isotopes and were not merely relative values. The results of the previously mentioned experiments, which were designed to measure and correct for discrimination effects due to gas flow, voltage scanning, variation in gas pressure, non-ohmic electrometer input resistors, impure sample gases, and instrumental drift, led the writers to assume that the resulting measurements were absolute.
Conclusion
Using the available literature, chromite ores gathered from various deposits throughout the world were assayed for the abundances of the chromium isotopes. However, no differences in the relative abundances were observed.
Upon application of criteria to determine the absoluteness of the measured abundances of the isotopes of chromium, the only discrimination observed during the measurements was that due to effusive gas flow out of the ionizing region of the ion source. The magnitude of this discrimination was determined from measurements of a mixture of separated nitrogen isotopes and corrections were made for it. On this basis, the abundances of the chromium isotopes reported in this communication can be considered absolute. Since chemical chromium has a common source, it is conservative to accept these absolute abundance values for the isotopes in reagent chromium. At the ±3σ level (99.7% confidence level), the recommended values in atoms percent are:
Cr50 = 4.352 ± 0.024
Cr52 = 83.764 ± 0.036
Cr53 = 9.509 ± 0.027
Cr54 = 2.375 ± 0.018
Using these values and the previously published and accepted values for the masses of the chromium isotopes [34,35], a value for the chemical atomic weight (conversion factor = 1.000275) of 51.998 ± 0.001 has been obtained wherein the stated uncertainty combines the effects of both mass and abundance measurements and is computed by conventional statistical methods.
References
- Aljerf L. Advanced highly polluted rainwater treatment process. J Urban Environ Eng. 2018; 12. Ref.: https://goo.gl/RKCzy2
- Aljerf L. High-efficiency extraction of bromocresol purple dye and heavy metals as chromium from industrial effluent by adsorption onto a modified surface of zeolite: kinetics and equilibrium study. J Environ Manage. 2018; 225: 120-132. Ref.: https://goo.gl/DehY8Y
- Nowak R, Konstantinov L, Hess P. Licvd of Cr(C,O) films from Cr(CO)6 at 248 NM: gas-phase and surface processes. Mater. Res Soc Symp Proc. 1988; 129: 85. Ref.: https://goo.gl/Q4rhA8
- Nier AO. A redetermination of the relative abundances of the isotopes of carbon, nitrogen, oxygen, argon, and potassium. Phys Rev. 1950; 77: 789-793. Ref.: https://goo.gl/rMbXpt
- Plies V. Mass Spectrometric investigations of the vapor phase over CrCl3 and CrCl3/Cl2. Cheminform. 1988; 19.
- Hibbs RG. Electron Microscopy of human apocrine sweat glands. J Investig Dermatol. 1962; 38: 77-84. Ref.: https://goo.gl/HKkoHH
- Bacuta GC, Kay RW, Rossman DL. High chromium and high aluminum deposits in the Zambales ophiolite complex, Luzon, Philippines: Origin and tectonic significance. Chem Geol. 1988; 70: 132. Ref.: https://goo.gl/XQo1KX
- Xibin W, Peisheng B. Genesis of Podiform Chromite deposits--evidence from the Luobosa Chromite deposits, Tibet. Acta Geol Sin-Engl. 2009; 61: 77-94. Ref.: https://goo.gl/LBCEzV
- Moutte J. Chromite deposits of the Tiebaghi ultramafic massif, New Caledonia. Econ Geol. 1982; 77: 576-591. Ref.: https://goo.gl/xNJRpH
- Bacuta GC, Kay RW, Gibbs AK, Lipin BR. Platinum-group element abundance and distribution in chromite deposits of the Acoje Block, Zambales Ophiolite Complex, Philippines. J Geochem Explor. 1990; 37: 113-145. Ref.: https://goo.gl/ywyLUY
- Verryn S. X-Ray powder diffraction data for Chromite from the UG-2 of the Bushveld Complex, South Africa. S Afr J Geol. 2008; 111: 225. Ref.: https://goo.gl/SQ8Cyh
- Prendergast MD. Archean Komatiitic sill-hosted chromite deposits in the Zimbabwe Craton. Econ Geol. 2008; 103: 981-1004. Ref.: https://goo.gl/KKU91k
- Page NJ, Engin T, Singer DA, Haffty J. Distribution of platinum-group elements in the Bati Kef chromite deposit, Guleman-Elazig area, eastern Turkey. Econ Geol. 1984; 79: 177-184. Ref.: https://goo.gl/m4UV2L
- Simonov VA, Ivanov KS, Smirnov VN, Kovyazin SV. Physicochemical parameters of the melts participating in the formation of chromite orehosted in the Klyuchevsky ultramafic massif, the Central Urals, Russia. Geol Ore Dep. 2009; 51: 109-122. Ref.: https://goo.gl/RzUx93
- Gazaleeva GI, Shikhov NV, Vlasov IА, Shigaeva VN. The Donskoy Ore Mining and Processing Industrial Complex chromite tailings retreatment technology development. Obogashch Rud. 2017; 1: 16-20. Ref.: https://goo.gl/WLv6y8
- Hammer S, Nettleton LL, Hastings WK. Gravimeter prospecting for chromite in Cuba. Geophys. 1945; 10: 34-49. Ref.: https://goo.gl/qLTPDX
- Guild PW. Petrology and structure of the Moa Chromite district, Oriente Province, Cuba. Trans. Amer Geophys Union. 1947; 28: 218. Ref.: https://goo.gl/G19iKP
- Mackowiak K, Pickles CA. Microwave reduction of Black Thor chromite ore. Can Metall Q. 2018; 57: 341-349. Ref.: https://goo.gl/9r15HB
- Agarwal S, Pal J, Ghosh D. Development of chromite sinter from ultra-fine chromite ore by direct sintering. Int Sci Int J. 2014; 54: 559-566. Ref.: https://goo.gl/3tSPNm
- Mondal SK, Mukherjee R. Chromite: Petrogenetic indicator to ore deposits. Ore. Geol Rev. 2017; 90: 63-64. Ref.: https://goo.gl/xRu2n4
- Jones VE. Chromite deposits near Sheridan, Montana. Econ Geol. 1931; 26: 625-629. https://goo.gl/zEdfCf
- Stobbe H. Chromite and other Minerals near Red Lodge, Montana. Rock Miner. 1962; 37: 117-124. Ref.: https://goo.gl/FqcsBx
- Flesch GD, Svec HJ. New preparation for chromyl fluoride and chromyl chloride. J Am Chem Soc. 1958; 80: 3189-3191. Ref.: https://goo.gl/esJTjf
- Flesch GD, White RM, Svec HJ. The positive and negative ion mass spectra of chromyl chloride and chromyl fluoride. Int J Mass Spectrom Ion Phys. 1969; 3: 339-363. Ref.: https://goo.gl/5tPj2i
- Duval C. Applied inorganic analysis (zéme edition). Anal Chim Acta 1953; 9: 390.
- Green PJ, Gard GL. Chemistry of chromyl fluoride. 5. New preparative routes to chromyl fluoride. Inorg Chem. 1977; 16: 1243-1245. Ref.: https://goo.gl/CFgi3a
- Sheft I, Martin AF, Katz JJ. High temperature fluorination reactions of inorganic substances with bromine trifluoride addition compounds1a,1b. J Amer Chem Soc. 1956; 78: 1557-1559. Ref.: https://goo.gl/E5Qe7F
- Young ED, Rumble D, Freedman P, Mills M. A large-radius high-mass-resolution multiple-collector isotope ratio mass spectrometer for analysis of rare isotopologues of O2, N2, CH4 and other gases. Int J Mass Spectrom. 2016; 401: 1-10. Ref.: https://goo.gl/MBh5m6
- Brodskii AI, Dontsova EI. Exchange between oxygen isotopes in inorganic solvents. Chem Abstr. 1940; 37: 4947.
- Mills GA. Oxygen exchange between water and inorganic oxy-anions. J Amer Chem Soc1940; 62: 2833-2838. Ref.: https://goo.gl/cejopN
- Lindau CW, Delaune RD, Patrick WH, Lambremont EN. Assessment of stable nitrogen isotopes in fingerprinting surface water inorganic nitrogen sources. Water Air Soil Pollut. 1989; 48: 489-496. Ref.: https://goo.gl/bwBvmR
- Russe K, Valkiers S, Taylor PDP. Synthetic isotope mixtures for the calibration of isotope amount ratio measurements of carbon. Int J Mass Spectrom 2004; 235: 255-262. Ref.: https://goo.gl/nKM7sf
- Junk G, Svec HJ. The absolute abundance of the nitrogen isotopes in the atmosphere and compressed gas from various sources. Geochim Cosmochim Act. 1958; 14: 234-243. Ref.: https://goo.gl/5eB2Ct
- Holden NE, Martin RL. Atomic weights of the elements 1981. Pure Appl Chem. 1983; 55: 1101-1118. Ref.: https://goo.gl/JEcm28
- Un A, Demir F. Determination of mass attenuation coefficients, effective atomic numbers and effective electron numbers for heavy-weight and normal-weight concretes. Appl Radiat Isot. 2013; 80: 73-77. Ref.: https://goo.gl/G9ZMYS