Review Article
Validation of HPLC-UV method for determination of amoxicillin Trihydrate in capsule

Sendanyoye Marcel*, Uwambajineza Tito, Ineza Ines and Nahimana Jean Pierre
University of Rwanda, College of Medicine and Health Science, Department of Pharmacy, Kigali, Rwanda
*Address for Correspondence: Sendanyoye Marcel, University of Rwanda, College of Medicine and Health Science, Department of Pharmacy, Kigali, Rwanda, Tel: +250783493980; Email: [email protected]
Dates: Submitted: 14 September 2018; Approved: 03 October 2018; Published: 04 October 2018
How to cite this article: Marcel S, Tito U, Ines I, Pierre. Validation of HPLC-UV method for determination of amoxicillin Trihydrate in capsule. Ann Adv Chem. 2018; 2: 055-072. DOI: 10.29328/journal.aac.1001014
Copyright License: © 2018 Marcel S, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Reverse phase; HPLC –UV; Counterfeit; Antimicrobial resistance; Buffer; Potency
Abbreviations: AOAC: Association of Official Analytical Chemists; CBER: Center for Biologics Evaluation and Research; ECTD: Electronic Common Technical Document; FDA: Food and Drug Authority; HPLC-UV: High Performance Liquid Chromatography-Ultraviolet Detector; ICH: International Conference on Harmonisation; ISO/IEC: International Organization for Standardization and the International Electro Technical Commission; MQCL: Medicines Quality Control Laboratory; NDA: National Drug Authority; NF: National Formulary; RT: Retention Time; RSD: Relative Standard Deviation; RRT: Relative Retention Time; RSB: Rwanda Standard Board; SE: Standard Error of Intercept; SD: Standard Deviation of Intercept; SSFFC: Substandard, Spurious, Falsely Labelled, Falsified and Counterfeit

Abstract
The intention of the present work is to validate an easy, better and reasonable approach for estimation of amoxicillin trihydrate in tablet formulation by opposite segment(reverse phase) HPLC –UV with advanced conditions and parameters for habitual use in Rwanda well known board in pharmaceutical laboratory in order to check if no substandard or counterfeit amoxicillin has entered in our country that can result in antimicrobial resistance, treatment failure which can be a chief difficulty on public health. an easy, selective, precise, speedy, specific, and correct reverse phase HPLC UV-seen technique has been verified for the dedication of amoxicillin, in addition that is a cost-effective technique for the established method, monobasic potassium phosphate (KH2PO4) used as buffer and methanol and had been used as a mobile section in the ratio 95:5 respectively. The elution turned into finished in an isocratic mode at a go with the flow rate of 1.5ml/minute proposed method became demonstrated as according to ICH guiding principle refereeing additionally to USP necessities for amoxicillin capsule. linearity range of amoxicillin and was evaluated inside the variety of 20–160 g/ml. the correlation coefficient r2 changed into 0.9998 and the relative well known deviation between six replicates injection was always much less than 2%. The retention time was found 3.5±0.02. the high percentage of healing of amoxicillin is 100.6±4% indicates that the proposed method is exceptionally correct and precise trueness of with the trueness of 100.06±1.2% .the statistical evaluation proved that the demonstrated method is appropriate for analysis of amoxicillin as the majority drug and pharmaceutical formula with none interference from excipients .with the aid of considering the efficiency of the drug samples, all analyzed pattern were within the variety of 90-120 % of percentage of labeled amount, but the efficiency had been distinctive amongst samples. The have a look at located that no counterfeit, no substandard product turned into amongst all batches of amoxicillin samples throughout the c programming language of the look at.
Introduction
Amoxicillin1 is chemically (2s.5r.6r)-6- [(2r)-2-amino-2-(4-hydrowyphenyl)-acetyl] amino-three, 3-dimethyl-7-oxo-four-thia-l-azabicyclo [3.2.0] heptane-2-carboxylic acid. It is freely soluble in water. The molecular components is C16H19N3O5S and molecular weight is 419.45 [1]. It is reliable drug in indian pharmacopoeia [2], British pharmacopoeia [3]. Amoxicillinis a mild– spectrum bacteriolytic β-lactum antibiotic used to deal with bacterial infections because of susceptible microorganisms. It is also the drug of preference inside the elegance due to the fact it’s far better absorbed following oral management. Amoxicillin acts by way of inhibiting the synthesis of bacterial cellular wall. It is soluble in water, methanol, barely soluble in ethanol and in part insoluble in toluene [4].
Literature survey
Well-knowns how’s that, handiest few spectrophotometric techniques and few analytical strategies were suggested for the quantitative estimation of amoxicillin in bulk drug and pharmaceutical formula. Consequently, a strive has been made to broaden new HPLC methods for its estimation in bulk and pharmaceutical formulation with accurate precision, accuracy, linearity and reproducibility (Figure 1).
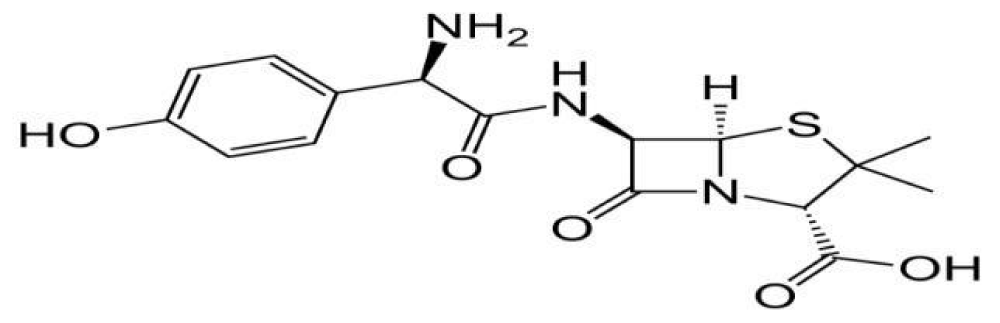
Figure 1: amoxicillin chemical structure.
Amoxicillin is an antibiotic beneficial for the remedy of a number of bacterial infections [5]. It’s far the primary line treatment for center ear infections. It could also be used for strep throat, pneumonia, pores and skin infections, and urinary tract infections amongst others. It’s miles taken with the aid of mouth [5].
Background of the problem
Antimicrobial resistance is a growing problem in developing countries and antibiotic use is sizeable. Most antibiotic use turned into in all likelihood needless or ineffective. There may be considerable problem approximately the high-quality of antimalarial capsules, with up to 35% terrible pleasant antimalarial suggested to be in malaria-endemic nations. But, little is known approximately the satisfactory of antibiotics, regardless of being in excessive call for globally. Producers of “counterfeit” [11], eleven drugs target economically profitable drugs, as well as people who have high extent sales. Over a length of 5 years, 2006–2010, 1.34 billion antibiotic prescriptions were disbursed within the USA [12] information are restrained from the growing global where first-line antibiotics may be easily obtained without prescription from pharmacies, grocery shops, or even cellular drug peddlers [13,14].
Substandard, spurious, falsely classified, falsified and counterfeit (SSFFC) scientific merchandise are by way of their very nature difficult to discover. They’re frequently designed to seem identical to the real product and won’t motive an apparent unfavorable response, however they regularly will fail to correctly treat the sickness or circumstance for which they were intended.
In 2013 who launched a worldwide surveillance and tracking gadget to inspire member states to document SSFFC incidents in a based and systematic layout, to assist increase a greater correct and demonstrated evaluation of the scope, scale and harm as a result of this difficulty. Over 920 medical merchandise have thus far been said representing all foremost healing categories and representing each innovator and conventional medicines.
The geared up availability of negative-exceptional drugs in growing countries results in remedy failure and, therefore, excess mortality and morbidity. Moreover, the great availability of substandard drugs plays a key position in growing the resistance to antimicrobial drugs [23]. We all understand that the primary aim of the pharmaceutical drugs is to serve the human to cause them to loose from potential infection or prevention of the disorder. For the drugs to serve its meant purpose they have to be loose from impurity or other interference which may damage human being. further of those, for the drugs to serve its meant cause it must include active pharmaceutical element in ordinary special limit of pharmacopoeia this means that it ought to not be a substandard medication or a counterfeit medication as we realize falsified scientific merchandise may incorporate no active component, the wrong active factor or the wrong quantity of the precise energetic aspect (who) [17], subsequently we need to make sure. Many contributors that produces capsules do now not have ok drug best manage for their facilities in their very own. For tablets imported in Rwanda or Rwanda local manufacturers’ batch certificates issued according with the Rwanda preferred board certification scheme so that it will generally offer sufficient records on the exceptional and beginning of a product. This assumes that a respectable inspection of the producing web site has been executed and that the manufacturer complies with accurate manufacturing practices. This implies that the consequences of an inspection, trying out by way of the competent country wide authority like Rwanda widespread board (RSB) need to display that the manufacturer is able to reliably producing a product of the desired high-quality. However, in positive conditions a want may additionally get up for national authorities to check samples whilst testing centers aren’t to be had. This trying out of excellent of some pharmaceuticals located in Rwanda retail pharmacies ought to show the overall evaluation on the excellent of drug treatments which can be being utilized by Rwandan population and to make certain that those drugs are of properly first-class. those test are essential due to the fact most of the time there are prolonged infection that are being determined at special hospitals for patient with distinctive infections and maximum physicians are claiming that there’s an opportunity that the antibiotics which can be being taken by means of their patients are substandard by suspecting that the to be had batch of antibiotic have negative first-rate which ought to result in antibiotic resistance. Tens of millions of tests, measurements and examinations are made each day in heaps of laboratories around the sector. There are innumerable reasons underpinning them, for instance: as a manner of valuing goods for trade purposes; supporting healthcare; checking the pleasant of drinking water, meals and feed; reading the basic composition of an alloy to verify its suitability for use in plane production; forensic analysis of frame fluids in crook investigations. Honestly each issue of society is supported in some way with the aid of analytical work. The value of carrying out these measurements is high and further fees might also arise from selections made on the idea of the consequences. for example, checks displaying capsules to be not worthy for intake may result in repayment claims; checks confirming the presence of banned capsules may want to result in fines, imprisonment or even, in a few international locations, execution. virtually it’s far essential to make an accurate measurement and be able to show that the end result is accurate, that is why the method that are being utilized in the ones measurements have to be confirmed and a tested technique will subsequently lead higher estimation of exact amount of energetic element found in drug and could help sooner or later to recognize the great of drugs especially antibiotic like amoxicillin in which this research is being emphasized. substandard drug and counterfeit drugs can also reason damage to patients and fail to treat the illnesses for which they had been intended, as a result they cause loss of self-assurance in medicines, healthcare carriers and health device(who).substandard and counterfeit medicines are by their nature difficult to discover, they are regularly designed to appear same to the real product and won’t motive an obvious destructive drug reaction, however they regularly fail to correctly treat the disease or condition for which they have been intended. the outcome, substandard of amoxicillin product ought to purpose resistance to microorganism but in case its satisfactory has been demonstrated to be in distinct restrict, if there’s antibiotic resistance inside the vicinity, the purpose has to be evaluated someplace else like overuse of antibiotic or other motive of antibiotic resistance. for the case of Rwanda, maximum medical practionners, pharmacist, nurse and patients are claiming to the first-class the drug which are getting used of their respective are however, their declare isn’t scientifically supported seeing that in previous years in our country the first-class of drug treatments changed into was evaluated bodily without any laboratory drug analysis to ensure the high-quality of purchased drug, thankfully given that 2015, pharmaceutical laboratory turned into hooked up at Rwanda standard board, therefore soon the claim from human beings, medical practionners, pharmacist or any other declare of bad fine changed into resolved by means of trying out of all medicinal drug fabricated from outdoor the country or produced domestically due to the fact most of the time the pleasant have to be ensured most effective with the aid of certificates of evaluation from outdoor which some time will be falsified.
This have a look at aimed to evaluate the fine of amoxicillin advocated for remedy of various infections that is domestically available within the Rwanda.
Analytical methods development
An analytical procedure is advanced to test a defined function of the drug substance or drug product against hooked up reputation standards for that characteristic. Early in the improvement of a brand new analytical manner, the selection of analytical instrumentation and methodology need to be decided on based totally on the intended reason and scope of the analytical method. Parameters that may be evaluated for the duration of approach improvement are specificity, linearity, limits of detection (LOD) and bounds of quantification (LOQ), range, accuracy, and precision [18].
Content of analytical procedures
You ought to describe analytical tactics in sufficient element to allow a ready analyst to breed the important situations and acquire outcomes in the proposed popularity standards. You should additionally describe elements of the analytical approaches that require special attention. An analytical system can be referenced from FDA-diagnosed resources (e.g., USP/NF, affiliation of analytical groups (AOAC) worldwide) if the referenced analytical method isn’t modified past what is allowed inside the posted approach. You must offer in element techniques from other published resources [18].
Analytical method validation
Validation is the affirmation by way of examination and the availability of objective proof that the precise requirements for a selected intended use are fulfilled (ISO/IEC 17025:2005 cl. 5.4.5.1). or a method of comparing method performance and demonstrating that it meets a specific requirement laboratories involved within the analysis of authentic samples should work in accordance with internationally permitted methods or standards-primarily based overall performance requirements and use of strategies of analysis that have, as far viable been, proven [12].
Why is method validation necessary?
Approach validation is vital for the motives below [12]
i. to ensure that the test technique give “accurate” results
ii. goal proof for defense in opposition to challenges
iii. clients need to be assured of the correctness of effects
iv. to provide dependable and accurate effects
v. to provide comparable consequences between laboratories
vi. to satisfy accreditation requirements
When to do method validation studies:
a. When considering purchasing a new system
b. When placing a new system into service
c. At regular intervals to assess on-going system performance
Verification
Verification is the confirmation, through the provision of objective proof that unique necessities were fulfilled (ISO 9000:2005 section 3.8.4) verification is relevant only for trendy/prevalent techniques that have been tested earlier than most often the vital parameters are the trueness and the precision [13].
a. non compendial analytical procedures
Analytical approach validation is the process of demonstrating that an analytical technique is suitable for its intended purpose. The technique and objective of the analytical procedures must be truly defined and understood before beginning validation research. This information is acquired from scientifically-primarily based technique improvement and optimization research. Protocols for both drug substance and product analytes or combination of analytes in respective matrices need to be evolved and performed. You ought to encompass information of the validation research and outcomes along with your software [18].
b. compendial analytical approaches
The suitability of an analytical method (e.g., USP/NF, the legit techniques of evaluation of AOAC global, or different identified trendy references) ought to be established below real conditions of use records to demonstrate that USP/NF analytical procedures are appropriate for the drug product or drug substance need to be blanketed in the submission and generated beneath a verification protocol [20].
The verification protocol should include, but is not limited to:
(1) compendial technique to be demonstrated with predetermined attractiveness standards, and
(2) details of the method (e.g., suitability of reagent(s), device, factor(s), chromatographic situations, column, detector kind(s), sensitivity of detector sign response, gadget suitability, sample preparation and stability).
The technique and quantity of verification should dictate which validation characteristic assessments need to be covered inside the protocol (e.g., specificity, LOD, LOQ, precision, accuracy). Issues that can affect what feature exams need to be inside the protocol may additionally rely on conditions including whether specification limits are set tighter than compendial attractiveness criteria, or RT or RRT profiles are changing in chromatographic techniques due to the fact of the synthetic direction of drug substance or variations in production technique or matrix of drug product. Robustness research of compendial assays do no longer want to be blanketed, if strategies are accompanied without deviations.
Statistical evaluation and models
Statistical analysis of validation facts may be used to assess validation characteristics in opposition to predetermined attractiveness criteria. All statistical processes and parameters used inside the evaluation of the information have to be primarily based on sound standards and appropriate for the intended assessment. several statistical strategies are useful for assessing validation traits, as an example, an evaluation of variance (ANOVA) to assess regression evaluation r (correlation coefficient) and r squared (coefficient of determination) or linear regression to degree linearity.
Many statistical methods used for assessing validation characteristics depend on populace normality, and it’s miles important to determine whether or no longer to reject this assumption. There are numerous strategies, inclusive of histograms, normality checks, and opportunity plots that may be used to evaluate the determined distribution. It is able to be suitable to transform the statistics to better fit the normal distribution or follow distribution-loose (nonparametric) methods when the found information is not commonly allotted. appropriate literature or text have to be consulted for records on statistical strategies to apply while growing new take a look at methods, evaluating current check strategies or evaluating size gadget overall performance, in addition to different fashionable statistics on the interpretation and treatment of analytical information. The statistics evaluation must be confident either by means of the usage of accurately confirmed software program or independent verification for correctness [21].
Revalidation
Principles described inside the validation follow to revalidation. When an exchange is made to an analytical method (e.g., an alternate in a bit of gadget or reagent or due to a trade in production procedure or formulation), revalidation of all or a part of the analytical system ought to be taken into consideration. Analytical method revalidation will also be warranted due to manufacturing system modifications, such as an alteration inside the drug substance production system that could affect technique performance (e.g., path of synthesis, fermentation) or advent of a brand new drug product system. You must revalidate to ensure that the analytical technique continues its critical performance traits (e.g., specificity, precision, accuracy). The degree of revalidation depends on the character of the change. In case of compendia approach revalidation can be essential inside the following instances: submission to USP of a revised analytical method or the usage of an established general approach with the new product or uncooked cloth. ICH documents deliver steerage on the necessity for revalidation inside the following instances: changes in synthesis of the drug substance, adjustments inside the composition of the drug product, and changes in analytical process [22].
Validation parameters or characterization of methods of analysis
Accuracy, specificity, selectivity, limit of detection, limit of quantification, linearity, recovery, repeatability, sensitivity.
Sensitivity: The change in response on a measuring instrument divided by the corresponding change in stimulus
Specificity: The ICH documents define specificity as the ability to assess unequivocally the analyte in the presence of components that may be expected to be present, such as impurities, degradation products, and matrix components [22,25].
Limit of detection: The detection limit of an individual analytical procedure is the lowest amount of analyte in a sample, which can be detected but not necessarily quantitatively determined as an exact value. Based on the standard deviation of the response and the slope, detection limit may be expressed as follow [25].
(LOD) may be expressed as: LOD= 3.3
where,
&= the standard deviation of the response for the lowest concentration in the range
s = the slope of the calibration curve.
Limit of quantitation: The quantification limit of an individual analytical procedure is the lowest amount of analyte in a sample, which can be quantitatively determined with suitable precision and accuracy. Based on the standard deviation of the response and the slope, the quantitation limit (LOQ) may be expressed as
LOD=10
where:
&= the standard deviation of the response for the lowest concentration in the range
s = the slope of the calibration curve.
I. Linearity and range: Linearity: the linearity of an analytical procedure is its ability to elicit test results that are directly, or by a well-defined mathematical transformation, proportional to the concentration of analyte in samples within a given range. Thus, in this section, “linearity” refers to the linearity of the relationship of concentration and assay measurement.
II. Range: the range of an analytical procedure is the interval between the upper and lower levels of analyte (including these levels) that have been demonstrated to be determined with a suitable level of precision, accuracy, and linearity using the procedure as written. The range is normally expressed in the same units as test results (e.g., percent, parts per million) obtained by the analytical procedure.
III. Determination of linearity and range: linearity should be established across the range of the analytical procedure. It should be established initially by visual examination of a plot of signals as a function of analyte concentration of content. If there appears to be a linear relationship, test results should be established by appropriate statistical methods (e.g., by calculation of a regression line by the method of least squares). Data from the regression line itself may be helpful to provide mathematical estimates of the degree of linearity. The correlation coefficient, y-intercept, slope of the regression line, and residual sum of squares.
Measurement uncertainty: A parameter linked to the measurement result and characterizing the spread of the values that can reasonably be added to the measurement magnitude.
The accuracy: The accuracy of an analytical procedure is the closeness of test results obtained by that procedure to the true value. The accuracy of an analytical procedure should be established across its range.
1. Determination: in the case of the assay of a drug substance, accuracy may be determined by application of the analytical procedure to an analyte of known purity (e.g., a reference standard) or by comparison of the results of the procedure with those of a second, well- characterized procedure, the accuracy of which has been stated or defined.
Accuracy is calculated as the percentage of recovery by the assay of the known added amount of analyte in the sample, or as the difference between the mean and the accepted true value, together with confidence intervals.
The ICH documents recommend that accuracy should be assessed using a minimum of nine determinations over a minimum of three concentration levels, covering the specified range (i.e., three concentrations and three replicates of each concentration). Assessment of accuracy can be accomplished in a variety of ways, including evaluating the recovery of the analyte (percent recovery) across the range of the assay, or evaluating the linearity of the relationship between estimated and actual concentrations. The statistically preferred criterion is that the confidence interval for the slope be contained in an interval around 1.0, or alternatively, that the slope be close to 1.0. In either case, the interval or the definition of closeness should be specified in the validation protocol. The acceptance criterion will depend on the assay and its variability and on the product. Setting an acceptance criterion based on the lack of statistical significance of the test of the null hypothesis [22].
Precision: Degree of conformity between independent measurement results obtained under prescribed conditions.
Repeatability: Repeatability expresses the precision under the same operating conditions over a short interval of time. Repeatability is also termed intra-assay precision [25].
Robustness/ruggedness: The degree of independence of the method of analysis from minor deviations in the experimental conditions of the method of analysis.
Selectivity: The selectivity of a method of analysis refers to the degree to which the method of analysis is usable for determining the presence of specific analytical parameters in a complex mixture (matrix) without interference from other analytical parameters in the mix.
Identification tests
The identity test ensures the identity of the analyte.
Purity tests: ensure the analytical procedure performed allow an accurate statement of the content of impurities of an analyte (eg: heavy metals limit, organic volatile impurity limit)the ICH documents state that when chromatographic procedures are used, representative chromatograms should be presented to demonstrate the degree of selectivity, and peaks should be appropriately labeled.
Peak purity tests (e.g., using diode array or mass spectrometry) may be useful to show that the analyte chromatographic peak is not attributable to more than one component [22].
i. Determinants of medicine qualityIdentity: presence of active ingredient
ii. purity: not contaminated with potentially harmful substances
iii. potency: usually 90–110% of the labeled amount
iv. uniformity: consistency of color, shape, size
v. Bioavailability: interchangeable products?
vi. stability: ensuring medicine activity for stated period for the drug that do not meet determinants of medicine quality, there several consequences that should be raised from using those drug such as [9] lack of therapeutic effects, prolonged illness, death, toxic and adverse drugs reactions, waste of limited financial resources, loss of credibility.
Research Methods
A study was carried out on amoxicillin purchased in the retail pharmacies of west, south, east and north province of Rwanda and Kigali town to find that there is a substantial amount of locally produced amoxicillin that are complying with drug monographs in the Rwanda. Pharmacopeia. the second stage is to detect the ingredients in each drug sample at a medicines quality control laboratory (mqcl) using the technique of high-performance liquid chromatography (HPLC) to then quantify the amount of active pharmaceutical ingredients. HPLC is regarded as the gold standard for drug quality analysis as it offers accuracy, specificity, and precision in quantifying the amount of stated active pharmaceutical ingredient detected or its absence.
Study design
Normally, this study is experimental qualitative and quantitative analysis for quality control purpose. Ten samples of amoxicillin capsules purchased from retail pharmacies, where 8 different batches were purchased randomly and at least in each province, we purchased 1 sample and in kigali we purchased 4 samples and analyzed for their drug content by a validated chromatographic method in order to verify if they complied with pharmacopeia requirements.
Material and equipment
i. volumetric flasks
ii. HPLC cecil
iii. detector: adept series: uv/vis detector (adept cecil ce 4300) at 254nm
iv. pump: adept series: dual piston pump (adept cecil ce 4900)
v. column hypersil reverse phase, c-18 column 250 mm x 4.6 mm i.d. particle size 5μm)
vi. software package: power stream
vii. ph meter(rsb/nqtl/pm001), sartorium pb-11
viii. analytical balance with the precision ± 0.0001g
ix. sonicator (rsb/nqtl/sn001) bandelin
x. beaker
xi. analytical pipette
Reagents and solution
a. pure standard of amoxicillin 86.2%(amoxicillin)
b. water HPLC grade used as solvent for diluent preparation
c. potassium dihydrogen phosphate (ec label2319134,ec-emb45053)
d. methanol hplc grade 99.8 (chem lab, belgium)
e. potassium hydroxide 45%(fishcer scientific ukltd, uk)
f. sulfiric acid ,analytical reagent,specific gravity of 1.84 at 25 0c(rfcl ltd, inde)
g. amoxicillin sample (Table 1)
| Table 1: Sample from different retails pharmacies of different province of Rwanda. | |||||
| designation batch expiry industry date | |||||
| 1 | clamoxicill® | 500mg | 2270 | jan-19 | glaxosmith,france |
| capsule | |||||
| 2 | clamoxicill® 500mg caps | 2096 | dec-17 | glaxosmith,france | |
| 3 | amoxicillin 500mg capsule | s365406 | 3/5/2018 | dawa ltd,kenya | |
| 4 | amoxicillin 500mg capsule | mx149 | feb-17 | sparsh biotech,inde | |
| 5 | amoxicillin 500mg capsule | 1510012 | sep-18 | dawa ltd,kenya | |
| 6 | amoxicillin 500mg capsule | 153132157 | oct-18 | reyound | |
| pharmaceutical | |||||
| co.ltd,china | |||||
| 7 | amoxicillin 500mg capsule | 61008 | dec-18 | dawa ltd,kenya | |
Diluent preparation: 13.6 g of monobasic potassium phosphate (KH2PO4) was dissolved in 2000 ml of distilled water, with the concentration of 6,8g/l where the PH obtained was 4.5, the PH adjusted to 5.0 ± 0.1 by using a 45% (w/w) aqueous solution of potassium hydroxide.
Chromatographic situations
flow rate: 1.5 ml/min
detection wavelength: 254 nm
injection volume: 20µl
temperature: room temperature
The content of the mobile phase potassium dihydrogen phosphate and methanol in the ratio (95:05v/v). The mobile phase was filtered through 0.45 μm membrane filter and sonicated for 15 min. the flow rate of the mobile phase was being maintained at 1.5 ml/min. the column temperature was kept at ambient and the detection was carried out by uv-detector wavelength at 254 nm. The run time was set at 10 min and the volume of the injection loop to be used was 20 μl. Prior to injection of the drug solution, the column was equilibrated for at least 30 min with the mobile phase flowing through the system.
Procedure
Standard stock solution preparation: Stock solution of amoxicillin was prepared by dissolving 100mg of amoxicillin in100ml standard volumetric flask containing approximately 50 ml of mobile phase and the solution was sonicated for 20 min and then the volume was made up to the mark with mobile phase to obtain a concentration of 1000 μg/ml.
Working solution preparation: using 10ml volumetric flask, and the formula of c1v1= c2v2 subsequent dilutions of standard stock solution was made with mobile phase to obtain the concentration range of 20-160 μg/ml. the standard solutions prepared as above were injected into the 20 μl loop and the we recorded the chromatogram. The retention time of amoxicillin was found on chromatogram. The calibration curve was constructed by plotting concentration against peak area ratio. The linearity was checked on calibration curve by looking also the value of correlation coefficient r2. The amount of amoxicillin present in sample was calculated through the standard calibration curve.
Method validation for amoxicillin
The proposed method was validated according to the international conference on harmonization (ICH) guidelines [25,26].
Our method was validated for precision (inter-day and intraday), linearity, accuracy, trueness, repeatability, sensitivity, specificity/selectivity, limit of detection and limit of quantification in RSB environment with RSB equipment, reagents, and environment.
Analysis and Interpretation of Data
Linearity and range
The linearity of an analytical procedure as the ability (within a given range) to obtain test result which are directly proportional to the concentration (amount) of analyte in the sample. it was studied by analyzing 5 concentrations of the reference standard using 6 replicates injections within days. So 5 aliquots of standard solutions of amoxicillin were taken in different 100ml volumetric flasks and diluted up to the mark with mobile phase to make concentrations of 20 μg/ml, 40 μg/ml, 80 μg/ml, 100 μg/ml, 160 μg/ml respectively. Those solutions were injected into the hplc system and pick area recorded. A regression equation y = 1.651x + 5.946 has been generated where y and x represent peak area and concentrations, respectively. Correlation coefficient was r2 0.9997, the corresponding calibration curve is represented as figure 3 (not shown). The retention time of amoxicillin was found to be 3.5 ± 0.02 min (Table 2).
| Table 2: Showing correlation between concentration and peak area. | ||
| lineality data sheet | ||
| concentration(μg/ml) | mean peak area n=6 (±sd) | rsd% |
| 20 | 39 | 1.9 |
| 40 | 71 | 2 |
| 80 | 140 | 1.9 |
| 100 | 170 | 0.6 |
| 160 | 270 | 1.8 |
| r2 | 0.9998 | |
| regression equation: y = 1.6517x+5.8667 linearity range 20-160 μg/ml |
||
The table 2 and figure 2 represents the linear regression analysis of calibration curve, where it represents the slope of the equation with the value of 1.651, the intercept 5.8667 and correlation coefficient R2=0.9998. With the standard error of 1.37. The data obtained are statistically significant with p <0.05 (where p value=0.02) at 95% confidence interval (Tables 3,4).
| Table 3: Showing the regression analysis, with correlation coefficient, slope and intercept are obtained from linear regression. | ||||||
| summary output | ||||||
| regression statistics | ||||||
| multiple r 0.999882495 | ||||||
| r square 0.999765003 | ||||||
| adjusted r square 0.999686671 | ||||||
| standard error 1.601527049 | ||||||
| observations 5 | ||||||
| anova | ||||||
| df | ss | ms | f | significance f | ||
| regression | 1 | 32736.03 | 32736 | 12763.14 | 0.0000015 | |
| residual | 3 | 7.694667 | 2.56489 | |||
| total | 4 | 32743.73 | ||||
| Coefficients | standard error | t stat | p value | lower95% | higher95% | |
| intercept | 5.946667 | 1.371466 | 4.33599 | 0.022632 | 1.58205 | 10.3113 |
| x variable | 1.651667 | 0.01462 | 112.974 | 0.0000015 | 1.60514 | 1.69819 |
| Table 4: System suitability parameters of chromatogram for amoxicillin. | |
| parameter | value |
| tailing factor | 2.1 |
| retention time(min) | 3.5±0.02 |
| LOD | 1.579043 |
| LOQ | 4.784979 |
Precision
Repeatability interday and repeatability intraday: Precision of the system was evaluated by analyzing 6 replicates injections of standard concentrations and % RSD (relative standard deviation) value of peak area and retention time was calculated to ensure that if it is ≤2%, this was calculated to determine any intra-day and inter-day variation [24]. The precision was assessed on repeatability inter -day and intraday.
i. inter-day repeatability: inter-day precision of the developed method was determined by 3 replicate analyses at low concentrations of standard (20µg/ml), medium (40,80 and 100µg/ml) and high (160µg/ml) on same day and on consecutive day respectively. The precision was calculated as % RSD of measured concentrations for each calibration level.
ii. intra-day repeatability: inter-day precision of the developed method was determined by analyses at low concentrations of standard (20µg/ml), medium (40, 80 and 100µg/ml) and high (160µg/ml) in triplicate within day for short period interval. The precision was calculated as %RSD of measured concentrations for each calibration level (Tables 5,6).
Reliability of estimate for precision data 29 within confidence interval, in method validation, confidence intervals are also used to indicate the reliability of an estimate. Confidence intervals provide limits around the sample mean to predict the range of the true population of the mean. The prediction is usually based on probability of 95%. The confidence interval depends on the sample standard deviation and the sample mean.
where s is the sample deviation, is the sample mean, n is the number of individual data points, and z is constant obtained from statistical tables for z.
The value of z depends on the confidence level listed in statistical tables for z. for 95%, z is 1.96. hence by adopting for 95 % confidence interval as shown in our regression analysis (Table 3), hence we adopt to use z=1.96.
retention time: 3.5 ± 1.96 = 3.5±0.02× = 3.48 to 3.52
with n=6 at 95% confidence interval the calculated confidence interval indicates that the range of the true population of the mean for the retention time for amoxicillin is between 3.52 and 3.48 min the RSD of amoxicillin peak area in inter-day repeatability was 0.7 and in intraday repeatability RSD was 0.3 with n=6
peak area: 38.5 ± 1.96× =37 to 40 with n=6 at 95% confidence interval
The calculated confidence interval indicates that the range of the true population of the mean for the peak area for standard for amoxicillin 20 μg/ml is 37 to38 mas the RSD of amoxicillin peak area in inter-day repeatability was 1.8 and in intraday repeatability RSD was 0.6 (Tables 5,6).
| Table 5: Showing the repeatability intraday with good precision. | |||
| repeatability interday | |||
| lever of injections | injections no | retention time | peak area |
| replicate1 | 3.53 | 39.1 | |
| replicate2 | 3.53 | 39.4 | |
| replicate3 | 3.52 | 40.6 | |
| 20 μg/ml | replicate4 | 3.54 | 39.5 |
| replicate5 | 0.5 | 36.3 | |
| replicate6 | 3.51 | 36.1 | |
| Mean | 3.523333 | 38.5 | |
| SD | 0.013663 | 1.853645 | |
| iRSD | 0.387775 | 1.830847 | |
| lever of injections | injections no | retention time | peak area |
| replicate1 | 3.52 | 70.4 | |
| replicate2 | 3.52 | 71 | |
| replicate3 | 3.51 | 72.2 | |
| 40 μg/ml | replicate4 | 3.49 | 73.6 |
| replicate5 | 3.48 | 68.7 | |
| replicate6 | 3.47 | 68.5 | |
| Mean | 3.498333 | 70.73333 | |
| SD | 0.02137 | 1.983599 | |
| RSD | 0.610855 | 2.804335 | |
| replicate1 | 3.51 | 142 | |
| replicate2 | 3.5 | 138 | |
| replicate3 | 3.5 | 142 | |
| 80 μg/ml | replicate4 | 3.46 | 142 |
| replicate5 | 3.45 | 138.1 | |
| replicate6 | 3.45 | 140.2 | |
| Mean | 3.478333 | 140.3833 | |
| SD | 0.027869 | 1.937438 | |
| RSD | 0.80121 | 1.380106 | |
| SENDANYOYE Marcel, University of Rwanda, College of Medicine and Pharmacy, Department of Pharmacy. | |||
| Table 6: Showing the repeatability intraday with good precision. | |||
| repeatability intraday | |||
| lever of injections | injections no | retention time | peak area |
| replicate 1 | 1 3.53 | 39.1 | |
| 20 μg/ml | replicate 2 | 3.53 | 39.4 |
| replicate 3 | 3.52 | 40.6 | |
| Mean | 3.526667 | 39.7 | |
| SD | 0.005774 | 0.793725 | |
| RSD | 0.16371 | 0.6245 | |
| replicate 1 | 3.52 | 70.4 | |
| 40 μg/ml | replicate 2 | 3.52 | 71 |
| replicate 3 | 3.51 | 72.2 | |
| Mean | 3.516667 | 71.2 | |
| SD | 0.005774 | 0.916515 | |
| RSD | 0.164175 | 1.28724 | |
| replicate 1 | 3.51 | 142 | |
| 80 μg/ml | replicate 2 | 3.5 | 138 |
| replicate 3 | 3.5 | 142 | |
| Mean | 3.503333 | 140.6667 | |
| SD | 0.005774 | 2.309401 | |
| RSD | 0.1648 | 1.641754 | |
Accuracy
A study of accuracy will be conducted by means of recovery studies. Recovery studies were carried out at three different levels. The pre analyzed sample was spiked with 80%, 100% and 120% of mixed standard solution. The mixtures were analyzed by the proposed method. The study was carried out in triplicate. Accuracy study was carried out by adding a known quantity of drug to quality control samples and reanalyzing contents by proposed method to find % recovery. By using the formula below we have calculated the percent recovery
Acceptance criteria
The mean % recovery of the amoxicillin trihydrate at each level should be not less than 90 % and not more than 105. % (Table 7).
| Table 7: the recovery studies of proposed HPLC method (n=3). | ||||||||
| s/n | sample concentration μg/ml | concentration spiked lever % | found concentration μg/ml | mean | standard deviation | Rrsd | recovery%(accuracy) | |
| 1 | 1088.8 | 80 | 1167.6 | 99.5 | 0.03 | 0.06 | 99.5± 0.3 | |
| 2 | 1030 | 100 | 1135.53 | 105.2 | 0.1 | 0.09 | 105.4± 0.1 | |
| 3 | 1030 | 120 | 1147 | 97.3 | 0.5 | 0.59 | 97.3 ± 0.6 | |
| Mean; 100.6±4.6 | ||||||||
by considering the recovery percentage and trueness, the accuracy of this method ranges from 97.3±0.6 to 105.4±0.1% which finally results the mean %recovery for each lever of spike of 100.06±4% with measurement of uncertainty, with 95% confidence interval, 100.6 = 100.6 ± 4.6 this implies that, the calculated confidence interval indicates that the range of the true population of the mean for the accuracy for amoxicillin is between 96 and 105.2 % .
Trueness
where mean of test results obtained for reference sample μ is the “true” value given for reference sample. Thus, the best trueness we can get is 100% (Table 8). The negative bias means that the amount of recovered are less than the true value by calculating the bias bias =
Hence relative percentage of the bias in this measurement should be calculated as: as we remember that, the best bias we can get is 0 (in units of the analyte) or 0 % respectively.
with measurement of uncertainty, with 95% confidence interval,100.06 = 100.06 ± 4.6. this implies that, the calculated confidence interval indicates that the range of the true population of the mean for the accuracy for amoxicillin is between 95.45 and 104.5% with measurement of uncertainty, with 95% confidence interval, 100.06 = 100.06 ± 1.03 this implies that, the calculated confidence interval indicates that the range of the true population of the mean for the trueness for amoxicillin is between 98.97 and 101.03%
Limit of detection
The detection limit of an individual analytical procedure is the lowest amount of analyte in a sample, which can be detected but not necessarily quantitated as an exact value. Based on the standard deviation of the response and the slope, detection limit.
(LOD) may be expressed as:
where,
s = the standard deviation of the response for the lowest concentration in the range
s = the slope of the calibration curve.
loq values for amoxicillin was 1.579 μg/ml. the low value of lod indicates that the method is sensitive table 9.
Limit of quantitation
The quantitation limit of an individual analytical procedure is the lowest amount of analyte in a sample, which can be quantitatively determined with suitable precision and accuracy.
Based on the standard deviation of the response and the slope, the quantification limit (LOQ) may be expressed as
LOQ values for amoxicillin was 4.785 μg/ml. the low value of LOQ indicates that the method is sensitive table 9.
Specificity and selectivity
Specificity/ selectivity the specificity/selectivity of the approach became executed via in identification take a look at with colorimetric technique as indicated by using worldwide pharmacopoeia fifth version and using chromatographic selectivity by means of assessing unequivocally energetic pharmaceutical ingredients within the presence of others factors which can be purported to be gift.
Identity technique (identification check) displaying the specificity: The identification of the active pharmaceutical factor become completed in keeping with the worldwide pharmacopoeia fifth edition, 2015, where 2 mg of every sample become placed in a test-tube and moisten with 1 drop of water, and about 2ml of sulfuric acid (~1760 g/l) ts changed into delivered in every test tube, the content material turned into mixed by means of swirling; the answer remained almost colorless. After setting the tube in a water-bathtub for 1 minute, the dark yellow coloration was formed showing the presence of amoxicillin. However, the colour was now not the same in all samples because of amoxicillin attention that must vary among samples.
Assay
The proposed tested method became correctly applied to determine amoxicillin trihydrate in artificial aggregate. Consequences are given in table 9. No interference of the excipients with the analyte of interest seemed; as a result, the proposed approach is suitable for the habitual analysis of amoxicillin trihydrate in capsule in synthetic mixture.
Subsequently, 20 drugs of every batch, every containing 500 mg of amoxicillin weighed as it should be. An amount equivalent to one hundred twenty mg (zero.12001g) of amoxicillin was weighed appropriately and transferred to a hundred ml volumetric flask containing approximately 50 ml of cell segment. The content became sonicated for 20 min and extent changed into made on top of things with the cell segment.
The ensuing answer become filtered via a membrane filter. The solution acquired became then diluted with the mobile segment if you want to reap an awareness of one thousand μg/ml of amoxicillin. Sample solution changed into injected under the same chromatographic conditions and the chromatogram became recorded in triplicate. The quantity of amoxicillin present in capsule system become determined by using comparing the height region from the usual.
After validation of all parameters, as proven in preceding web page on essay to calculate the amount in microgram of amoxicillin per mg of amoxicillin in the portion of capsule the usage of linear regression equation.
This was completed through thinking about also the average amount of sample powder weighed for every capsule, extent of flask used, dilution of pattern carried out, the attention acquired on HPLC the usage of calibration curve, as a result percent content of label claim (efficiency) has been decided (Table 10).
The amount of amoxicillin for each formulation were determined by using the methods described in the USP 29 monograph for amoxicillin.
Amoxicillin capsule contains not less than 90% and not more than 120% of the labeled amount of amoxicillin USP volume.
Results of Findings and Discussion
Optimization of the chromatographic conditions was carried out with various combinations of buffer and methanol and by observing the peak parameters, the run time of the method was set at 10 min, the absence of interference peak with blank and sample at the retention time of amoxicillin (3.5minutes) ensured the specificity of the method, with RSD 0.7% (Table 5) which indicates a good base line. When the same drug solution was injected 3 times, the retention time of the drug was same. Linearity range was observed in the concentration range of 20-160 μg/ml (Table 2 and Figure 3). The regression equation of amoxicillin concentration over its peak area ratio was found to be y = 1.6517x+5.8667 (R2=0.9998) where y is the peak area ratio and x is the concentration of amoxicillin (Figure 2). The proposed HPLC method was also validated for intra-day and inter-day variation. the relative standard deviation in the peak area of the drug for 6 replicate injections in inter-day and 3 replicate injections in intraday was found to be less than 2% for all the standard peak area obtained Table 5,6 respectively. The tailing factor was found to be 2.1, which indicates good shape of peak. the limit of detection and limit of quantification was found to be 1.579µg/ml and 4.785µg/ml respectively (Table 9), which indicates the sensitivity of the method the use of buffer and methanol in the ratio of 95:05 v/v resulted in peak with good shape (sharp peak). The high percentage of recovery of amoxicillin is 100.6±4% indicates that the proposed method is highly accurate (Table 7) and good trueness of with the trueness of 100.06±1.2% (Table 8) no interfering peaks were found in the chromatogram indicating that excipients used in tablet formulation did not interfere with the estimation of the drug by proposed HPLC method.
| Table 8: The mean recovery indicating the trueness of the method. | |||||
| s/n | concentration μg/ml | average peak area | found concentration | recovery % | bias % |
| 1 | 20 | 39 | 20.02059358 | 100.103 | 0.102968 |
| 2 | 40 | 71 | 39.40278619 | 98.50697 | -2.98607 |
| 3 | 80 | 104.4 | 81.43791641 | 101.7974 | 7.189582 |
| 4 | 120 | 171 | 99.9721381 | 99.97214 | -0.13931 |
| 5 | 160 | 270 | 159.9357965 | 99.95987 | -0.32102 |
| Mean: 100.0679 SD: 1.167234 RSD: 1.166442 | |||||
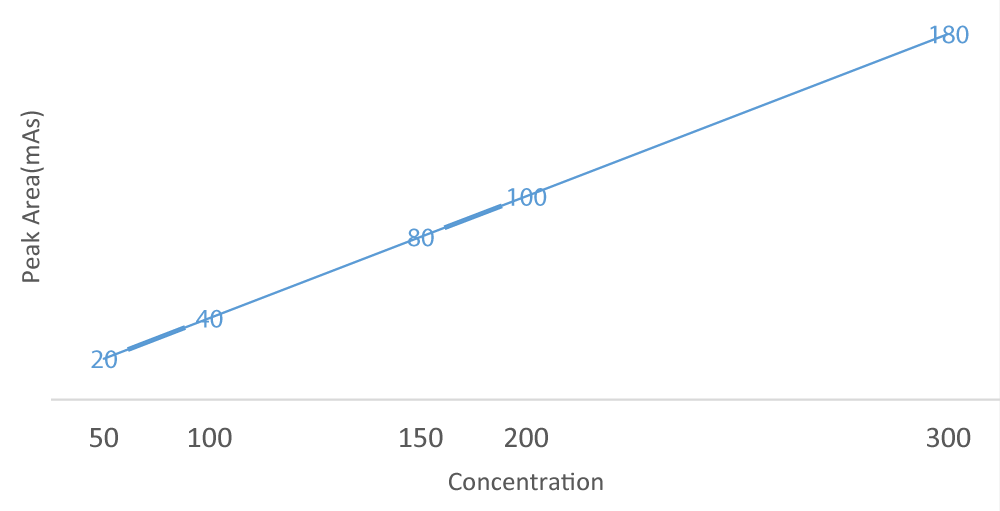
Figure 2: Showing correlation between concentration and peak area.
The relative standard deviation (RSD) of the chromatographic method was less than 2 % within a day and less than 2 % within three days, which complies with the usp [29] requirements (RSD should be less than 2%) and proving the precision of the method. the results of the drug content (Table 10) show that all formulations complied with usp [29] specifications for amoxicillin content (90% - 120% of the labelled content) containing 500 mg amoxicillin per capsule.
| Table 9: Regression analysis data and summary of validation parameters for proposed method. | |
| parameter | value |
| trueness | 100.06±1.3 |
| slope | 1.651 |
| intercept | 5.946 |
| linearity range | 20-160 μg/ml |
| retention time (min) | 3.53.5±0.02 |
| tailing factor | 2.1 |
| correlation coefficien(r) | 0.9997 |
| se of intercept | 1.371 |
| se of intercept | 3.2904 |
| SD of lowest conc in the range | 0.79 |
| LOD | 1.579043 |
| LOD | 4.784979 |
| recovery % | |
| level 1 | 99.5±0.3 |
| level 2 | 105.4±01 |
| level 3 | 97.3±0.6 |
| accuracy | |
| 1specificity | specific method |
| precision: | 0.5 |
| repeatability intraday % RSD n=6) | 0.4 |
| repeatability interday % RSD n=6) | 0.7 |
| sensitivity | very sensitive |
| iSENDANYOYE Marcel, University of Rwanda, College of Medicine and Pharmacy, Department of Pharmacy. | |
| Table 10: Table showing essay of amoxicillin sample. | |||||||
| S/N | Item Description | Industry | Batch | Mtg Date | Expiry Date | %Potency | Mean, Sd, Rsd |
| 1 | clamoxicill® 500mg caps | dawa ltd,kenya | 2270 | 16-jan | 19-jan | 108.9 0.02 0.02 |
mean sd rsd |
| 2 | clamoxicill® 500mg caps | sparsh biotech,inde | 2096 | 14-dec | 17-dec | 105.6 0.49 0.46 |
mean sd rsd |
The results obtained on amoxicillin method validation are similar to the results of Manzoor Ahmed, Suresh Babu g*, Sathish Kumar Shetty [27], estimation of amoxicillin trihydrate: absence of interfering peaks from excipients and drug content of all amoxicillin trihydrate sample was in the range of 91.06-108.9% of the labeled claim reflected that the developed method could be used for the determination of amoxicillin trihydrate capsule.
The results obtained on amoxicillin essay are similar to the results of Manzoor Ahmed, Suresh Babu G*, Sathish Kumar Shetty [27] and those found by prof kayumba pierre claver [28].
Conclusion and Recommendation
The validated HPLC method was found to be simple, selective, specific, rapid, sensitive, repeatable, precise and accurate for the estimation of amoxicillin in pharmaceutical formulations. In addition of this, the validated method is cheap (economical method) and friendly to environment because it uses methanol instead of acetonitrile in mobile phase. Hence, this method can easily and conveniently be adopted for routine quality control analysis of amoxicillin in bulk and pharmaceutical formulations.
The qualitative analysis of this method was found to be successful with dark yellow color characterizing the presence of amoxicillin in sample. Another qualitative aspect was peak identification on chromatogram of amoxicillin sample was at the same retention time as for amoxicillin standard. by ensuring the result of identification and an essay, no amoxicillin substandard or counterfeit was found among province of Rwanda pharmaceutical market; briefly all medicines assessed were of good quality either potency or physical identification.
Based on our findings we recommend: a systematic quality test of all essential drug formulations available on the Rwandan market, the registration of each commercially available drug, documenting its specifications, and most importantly the verification of these specifications. This method can easily and conveniently be adopted for routine quality control analysis of amoxicillin in bulk and pharmaceutical formulations in pharmaceutical quality control laboratory at Rwanda standard board.
References
- Ref.: https://goo.gl/qcv41u
- Indian pharmacopoeia. Ghaziabad, The Indian pharmacopoeia commission. 2003; 3: 2090-2095. Ref.: https://goo.gl/mKvK6a
- British pharmacopoeia, her majesty’s stationary office. London. 2003; 3: 719-721.
- http://msds.chem.ox.ac.uk/am Ref.: https://goo.gl/JPbfNA
- The american society of health-system pharmacists. retrieved 1 august 2015. Ref.:
- Basic principles of hplc ppt by martin r. hackman nj- depoffice of quality assurance Ref.: https://goo.gl/5NaEfz
- Koerner PJ. General principles of hplc method development, Thailand 2013: Ref.: https://goo.gl/huK1Ye
- Ref.: https://goo.gl/8LzS2M
- Chapter 18, “Quality assurance for drug procurement,” of managing drug supply 2nd ed. Management sciences for health and world health organization. 1997;
- The International Pharmacopoeia. 2003; 5: Ref.: https://goo.gl/6dmv18
- Ref.: https://goo.gl/YC1Hdq
- Schulze M. Transfer of validated methods into laboratories working routine.
- Thompson M, Stephen l, Ellison R, wood R. Harmonized guidelines for single-laboratory validation of methods of analysis (IUPAC Technical Report). in: pure appl chem. 2009; 74: 835–855, Ref.: https://goo.gl/eSn6L6
- Guidance for Industry. Ind meetings for human drugs and biologics, chemistry manufacturing and controls 529 information. 2001; Ref.: https://goo.gl/enMeZw
- Inds for phase 2 and 3 studies of drugs, including specified therapeutic biotechnology- 532 derived products. 1999;
- Investigating out of specification (oos) test results for pharmaceutical production. 2006; Ref.: https://goo.gl/gQt3ck
- Guidance for Industry. Process validation: general principles and practices. 18. u.s. 2011; Ref.: https://goo.gl/xkXUQ3
- Department of health and human services, food and drug administration center for drug evaluation and research (cder), center for biologics evaluation and research (cber) (july 2015 pharmaceutical quality/cmc)” analytical procedures and methods validation for drugs and biologics (guidance for industry)”
- Guidance for industry: international conference on harmonization)
- q1c stability testing for new dosage forms. 1997; Ref.: https://goo.gl/QBWFZP
- q2(r1) validation of analytical procedures: text and methodology. 1995;1997; Ref.: https://goo.gl/weJhxX
- Pharmacopoeia (usp 37). Validation of compedial methods.
- Prazuck T, Falconi I, Morineau G, Bricard-Pacaud V, Lecomte A, et al. Quality control of antibiotics before the implementation of an std program in northern myanmar. Sex Transm Dis. 2002; 29: 624-627 Ref.: https://goo.gl/tXbrzz
- Shabir GA. Step-by-step analytical methods validation and protocol in the quality system compliance industry. Ref.: https://goo.gl/xrc8kH
- International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use, validation of analytical procedures: text and methodology q2(r1), current step 4 version parent guideline dated 27 october 1994 (complementary guideline on methodology dated 6 november 1996 incorporated in november 2005).
- The international conference on harmonization, q2(r1), validation of analytical procedure, text and methodology. 2005; 1-13.
- Ahmed M, Suresh Babu G, Shetty SK. Development and validation of amoxicillin by rp-hplc method in bulk drug and pharmaceutical dosage forms, international journal of chemtech research coden(usa). 2011; 3: 1037-1041. Ref.: https://goo.gl/nPEQtX
- Claver KP. Quantitative drug analysis and evaluation of the influence of accelerated stability testing on the in vitro dissolution.
- Webster K. Statistical analysis in analytical method validation. 2013; Ref.: https://goo.gl/KGNMGm